Getting started with LAMMPS


What is LAMMPS?
LAMMPS is a classical molecular dynamics code with a focus on materials modeling. It’s an acronym for Large-scale Atomic/Molecular Massively Parallel Simulator.
LAMMPS has potentials for solid-state materials (metals, semiconductors) and soft matter (biomolecules, polymers) and coarse-grained or mesoscopic systems. It can be used to model atoms or, more generically, as a parallel particle simulator at the atomic, meso, or continuum scale.
Install LAMMPS:
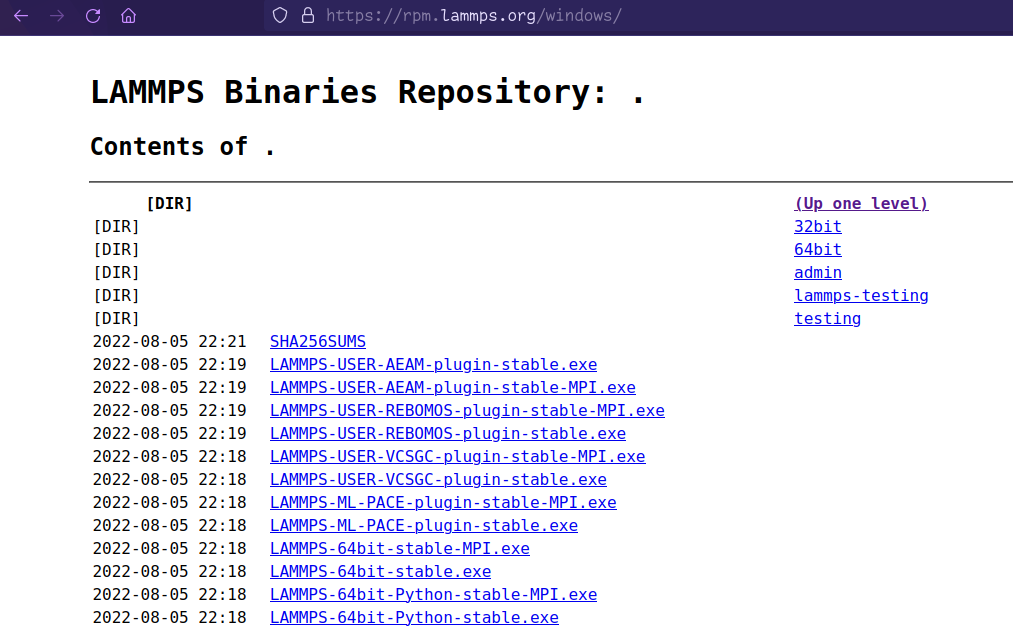
Windows: https://rpm.lammps.org/windows/
Here choose the LAMMPS-64bit-stable-MPI and download it:
Linux: https://docs.lammps.org/Install_linux.html
Now.... Let's download VScode.
Go here: https://code.visualstudio.com/
Download and install it.
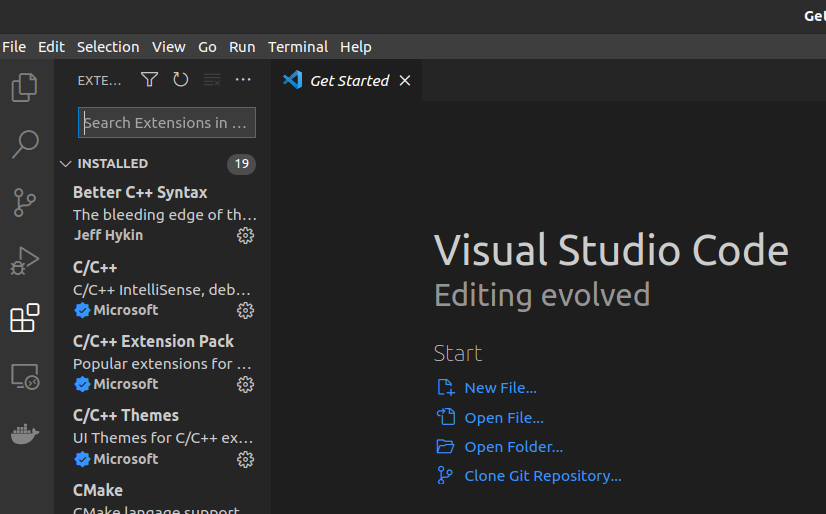
Then... Click on Extensions(The Rubik's cube like icon):
On the Search Box type: lammps
You'll get these two:
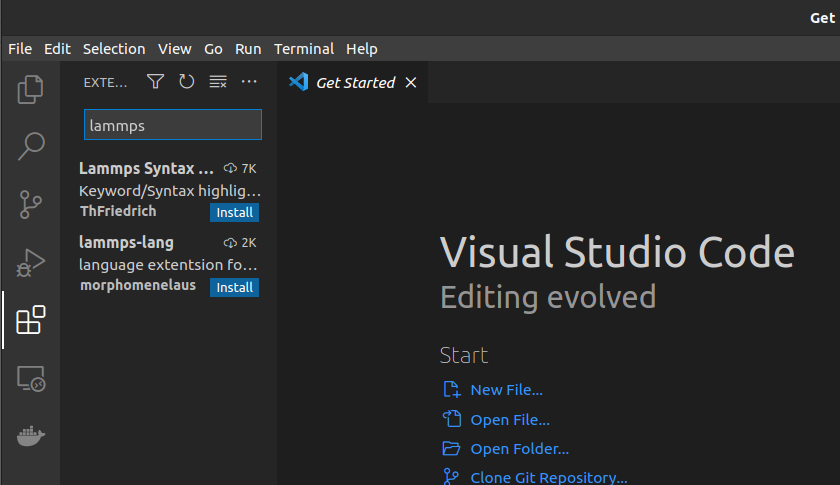
Install the first one which says: Lammps Syntax...
Now you're ready to fly!!!
What to do after these?
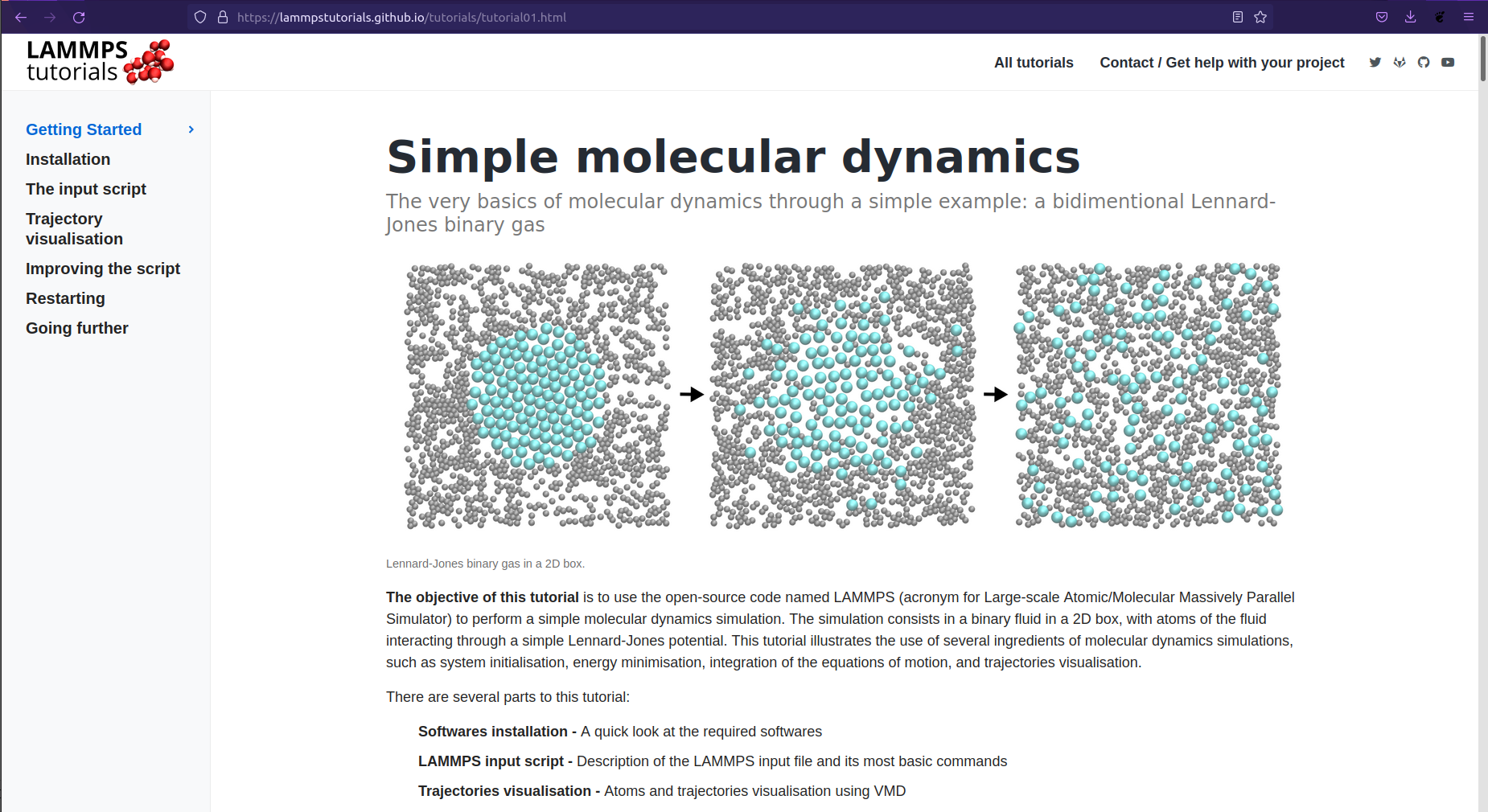
Take a long breath & go here: https://lammpstutorials.github.io/index.html

Choose one and click it.
Here I've chosen the first one:
Read the Getting Started and go to The input script section. Here we're skipping the installation step as we've already done that.
You'll see:
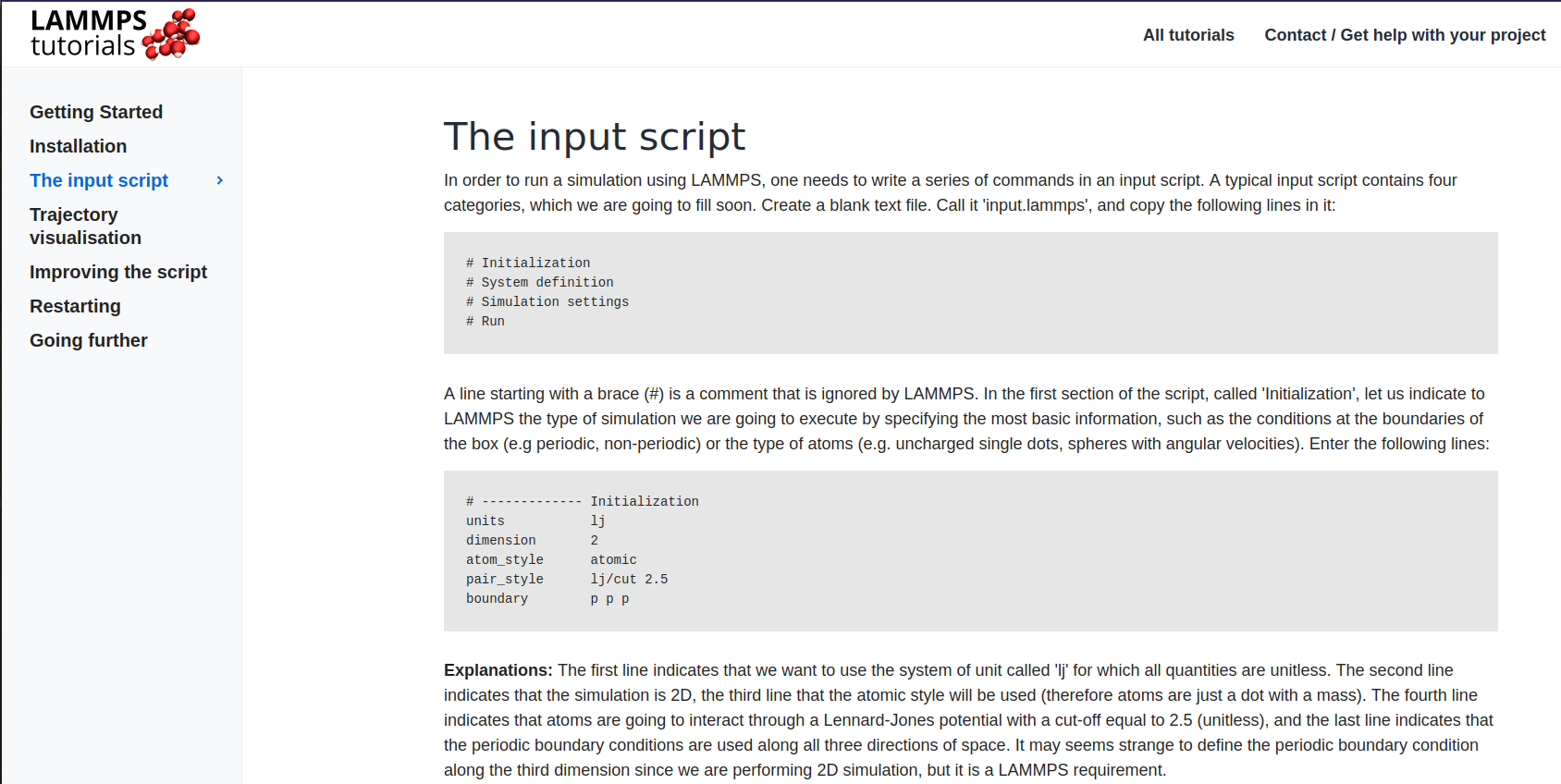
Here the grayed area has codes or commands. Commands are to be run on the LAMMPS shell or terminal of VScode.
Now... let's create a input.lammps file in VScode by clicking File on the upper left and choosing New File. Here I've created the file inside a folder titled "lammps". Now copy and paste the below lines:
# Initialization
# System definition
# Simulation settings
# Run
# ------------- Initialization
units lj
dimension 2
atom_style atomic
pair_style lj/cut 2.5
boundary p p p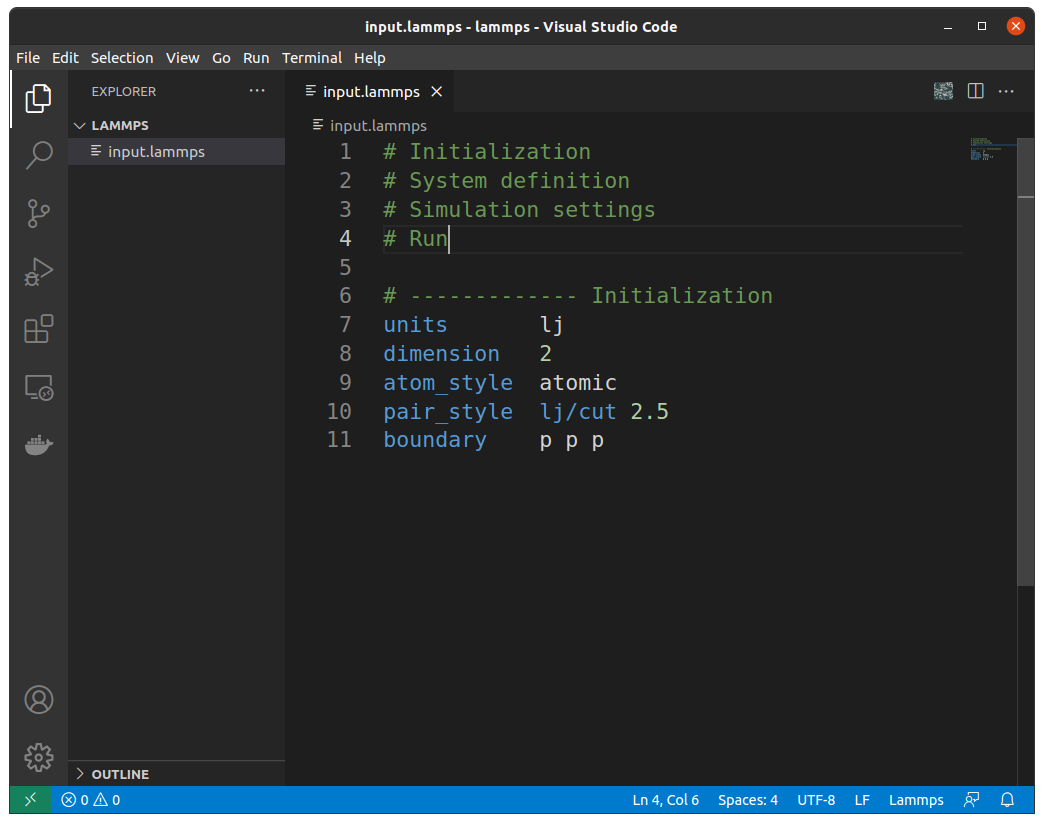
Each LAMMPS script has four steps:
a. Initialization
b. System Definition
c. Simulation Settings
d. Run
In this tutorial I'll only write the Initialization step.
If you hover on the code, for example here if I hover on units you'll see it's use and examples:
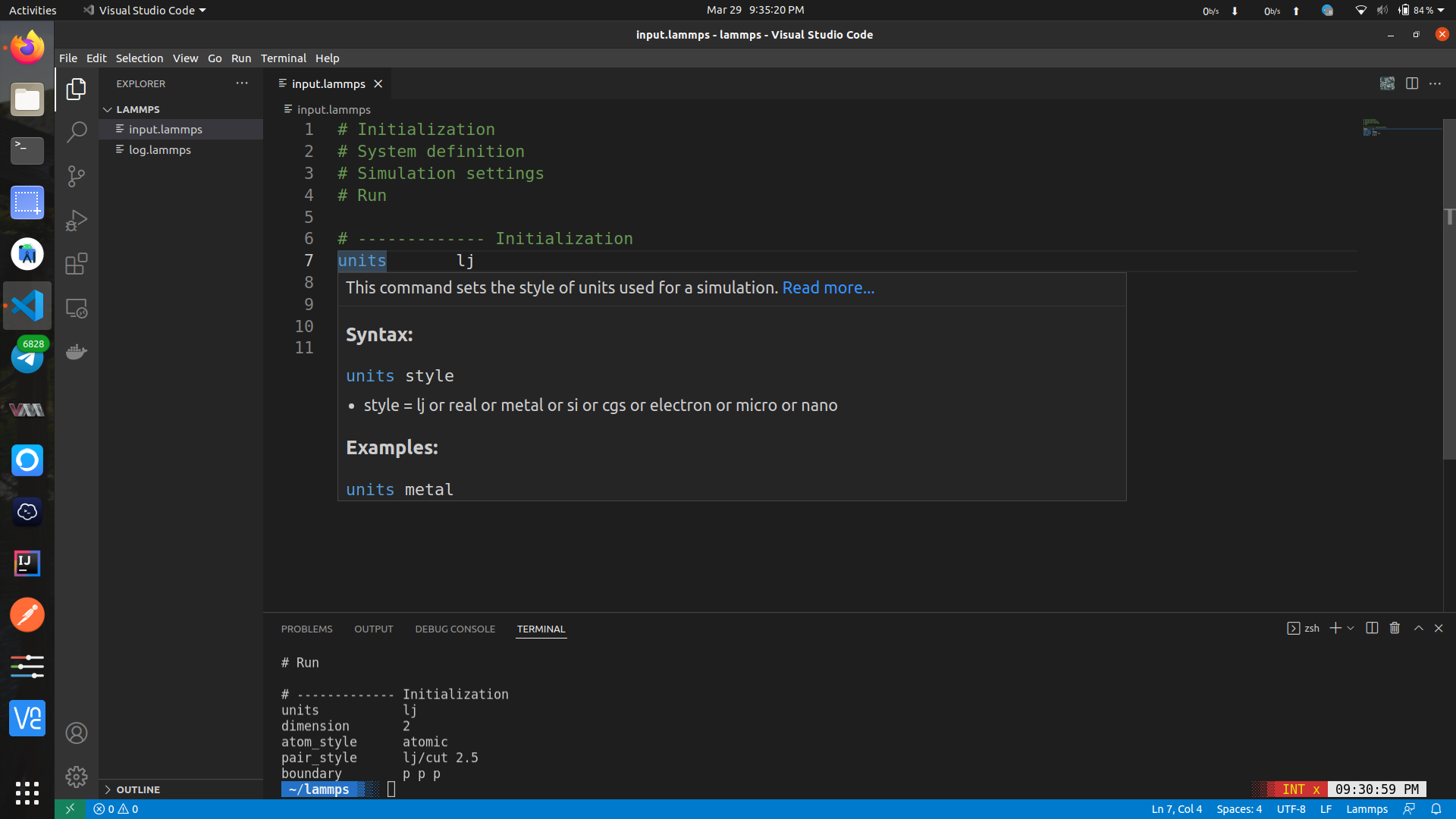
Now.... Let's open the terminal:
Click on the upper left Terminal or press ctrl + shift + `You'll get this in the bottom:
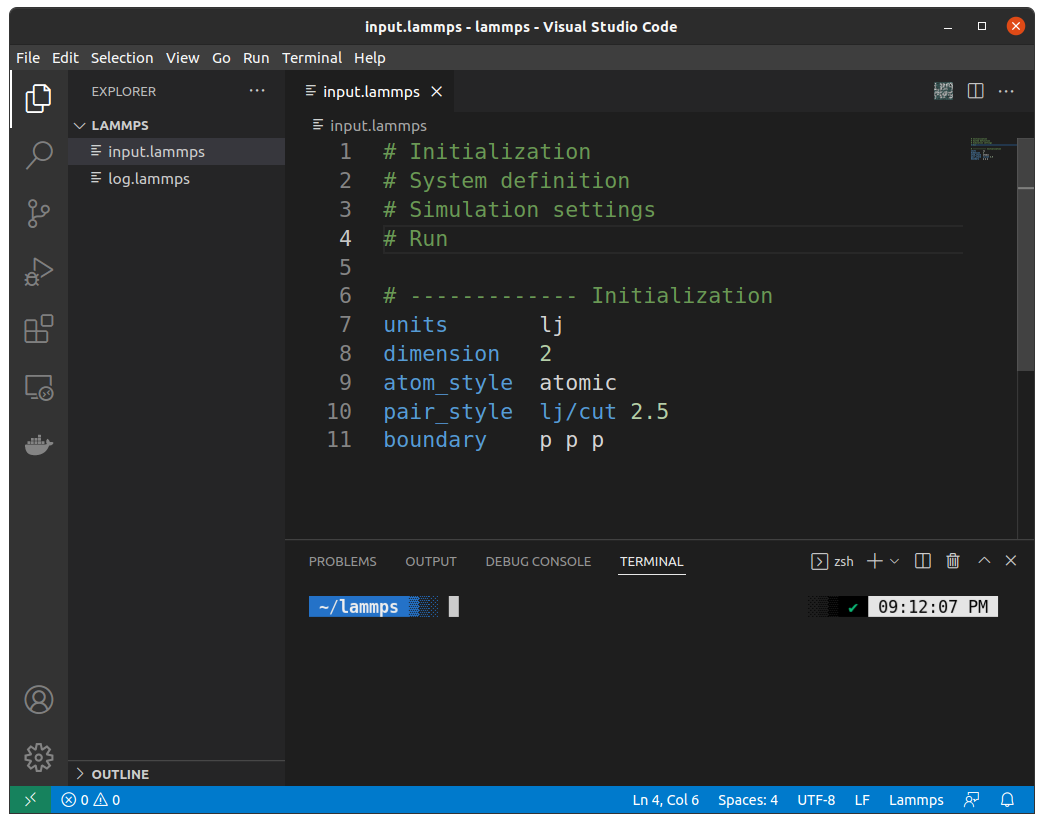
Type lmp here and hit enterIf you get something like below:
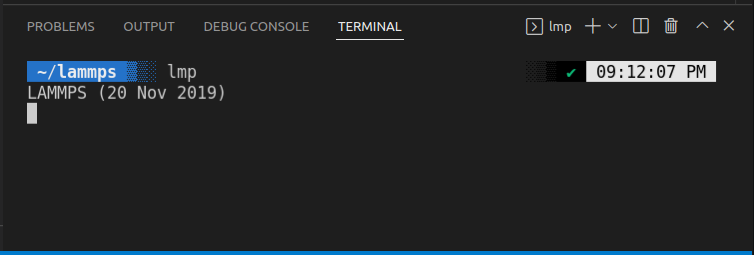
Then LAMMPS is succesfully installed. Else try reinstalling LAMMPS & try again.
Hit ctrl + c
Now type the below lines in the terminal and hit enter:
lmp -in input.lammpsYour terminal will look like this:
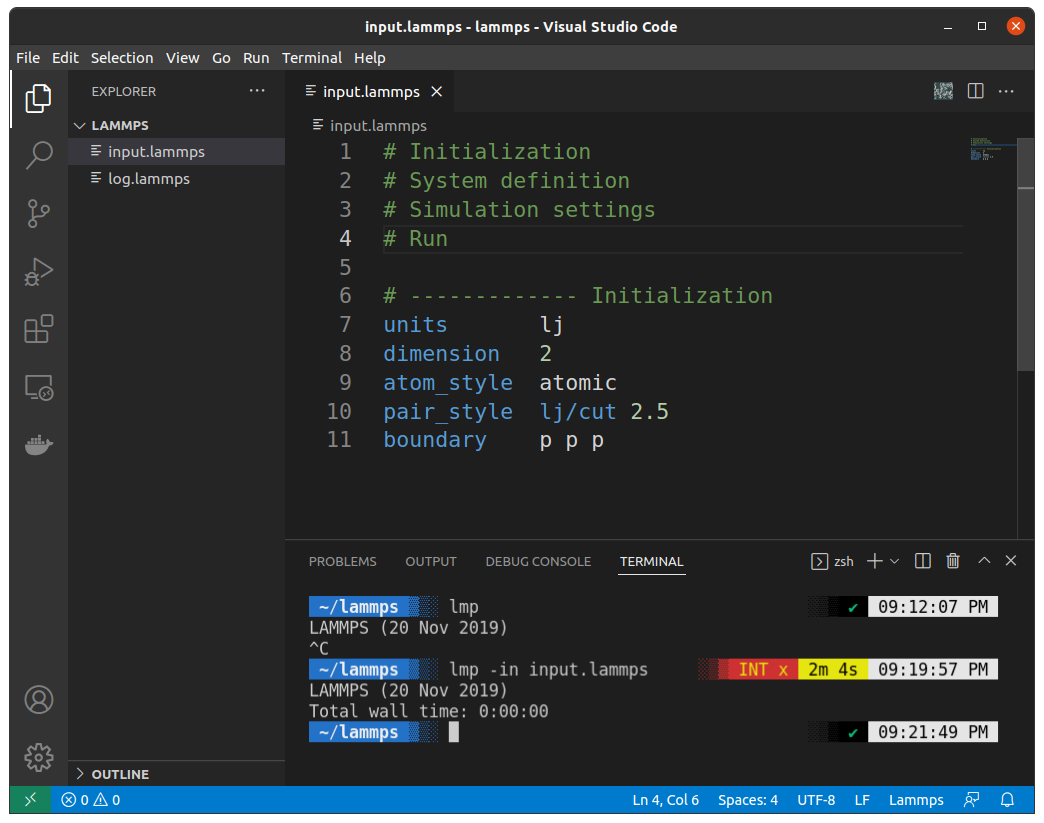
Congo!!! You've just run your first LAMMPS code!
Explore https://lammpstutorials.github.io/tutorials/01-SimpleMolecularSimulation.html to finish the code.
Examples:
1. https://lammpstutorials.github.io/index.html
2. LAMMPS documentation: https://docs.lammps.org/Manual.html
References:
Upcoming post: Visualizing the data with OVITO
Last update: October 3, 2022